by Ali Rind, Last updated: April 2, 2026

A sponsor monitoring visit is scheduled for next week. The request covers source documents for 30 subjects across three document types: patient notes, lab reports, and consent forms. Your team needs to prepare a redacted pack, verify it is complete, and deliver it through a secure channel, with a documented audit trail.
Most research sites have no standard process for this. Some redact manually in Adobe. Some apply black boxes without checking whether the underlying text is actually removed. Some miss fields and only discover the gap during the audit itself.
This guide covers the complete process for redacting clinical trial source documents before sharing with sponsors: what must come out, in what order, and how to document it in a way that stands up to scrutiny.
What Are Source Documents in a Clinical Trial and Why Sponsors Need Them
Source documents are the original records from which clinical trial data is derived. Under ICH GCP E6(R2), they include hospital records and patient notes, clinic charts, laboratory results, pharmacy records, medical imaging reports, and investigator observations.
Sponsors need access to source documents to verify that data entered on the Case Report Form (CRF) matches the underlying patient record. This process is called source data verification (SDV). Regulatory inspectors from agencies including the MHRA, FDA, and EMA may also request access during inspections.
The compliance challenge is straightforward. These documents contain direct patient identifiers. Sharing them with a sponsor's monitoring team or an external CRO creates obligations under GDPR and UK GDPR that require appropriate controls before transmission.
What Regulations Require Redaction Before Sharing
Three regulatory frameworks govern what must be protected when sharing clinical trial source documents externally.
-
ICH GCP E6(R2) requires that trial-related records are maintained in a way that protects patient privacy. Section 4.9 covers source documents and requires that they be accurate, complete, and protected from unauthorised access or modification.
-
GDPR and UK GDPR classify patient names, dates of birth, NHS numbers, and medical record numbers as personal data under Article 4. Sharing these with an external sponsor team requires either an appropriate lawful basis and data processing agreement, or de-identification to remove direct identifiers before sharing. The UK GDPR (post-Brexit) applies the same requirements for research sites based in the United Kingdom.
-
NHS Data Security Standards require NHS Trusts and NHS-run research sites to control access to patient identifiable information and maintain records of disclosure. The Data Security and Protection Toolkit outlines the operational requirements.
Together, these frameworks require that any personal data shared with external parties is pseudonymised or de-identified to remove direct identifiers before transmission. For organisations handling broader healthcare data compliance, VIDIZMO's healthcare data and AI solutions provide an overview of how these requirements can be addressed systematically across document types.
What Must Be Redacted: PII Checklist for Clinical Source Documents

The final item requires explanation. In a rare disease trial where only two subjects were dosed at a site on a given day, including the exact dosing date in a shared document effectively identifies both patients. The subject code is visible on the CRF. The date narrows it down to one person. In larger trials this risk is lower, but the principle applies: dates that are identifying in context must be treated as PII.
Not every redaction scenario calls for removing every identifier type. Where your protocol or data sharing agreement specifies a narrower scope, selective PII redaction allows you to target specific identifier categories without over-redacting data that is legitimately required for verification.
The Redaction Workflow Step by Step
A repeatable process for clinical trial document redaction covers six stages.
Stage 1: Receive and log the request. Record what has been requested, by whom, for which trial identifier, covering which date range, and for what purpose (monitoring visit, audit, inspection). This becomes the first entry in your audit trail.
Stage 2: Identify the relevant documents. Pull the correct files from your trial master file or electronic health record system. Confirm you are working with copies. Never modify originals. If your document management system does not support working on copies automatically, create them manually before opening any file.
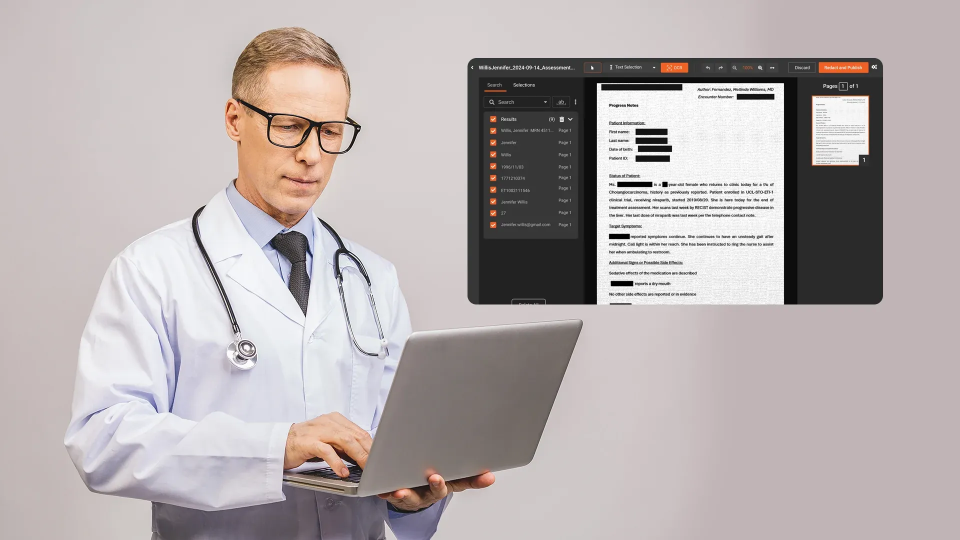
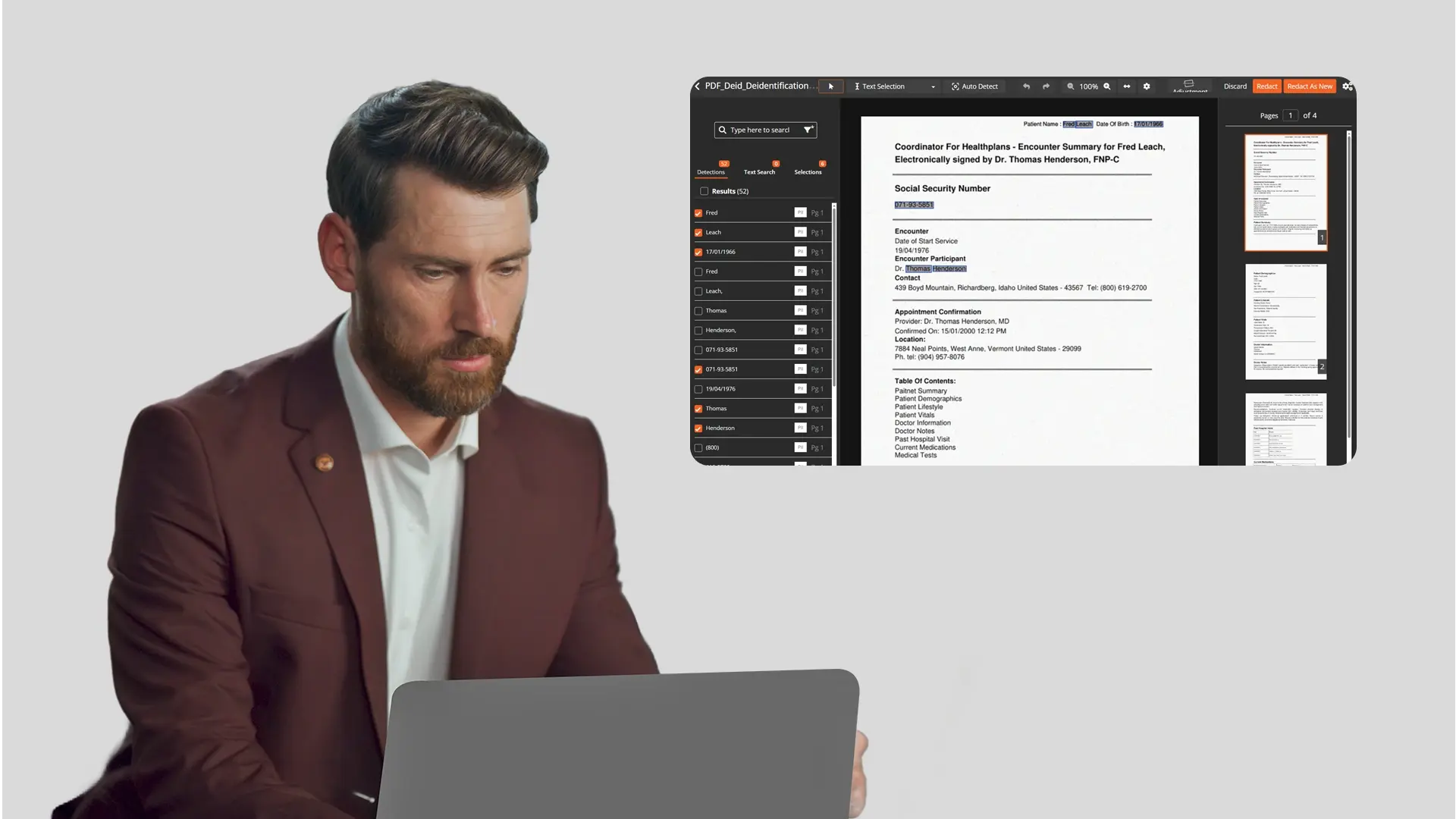
Stage 3: Run automated PII detection. Use a purpose-built redaction tool to scan each document for the identifiers listed above. For clinical source documents, detection must cover both structured fields (name and date fields in forms) and unstructured narrative text (identifiers embedded in physician notes and letters). Scanned documents require optical character recognition (OCR) before text-based detection can run. Handwritten content, common in clinical notes, requires intelligent character recognition (ICR). For a practical explanation of how OCR-based detection works in document workflows, see OCR-Based Text Redaction: A Practical Guide.
Stage 4: Human review. Before finalising, review the automated output. Check sections where identifiers commonly appear in non-standard positions: document headers, footers, watermarks, and free-text sections. Add manually identified items the AI missed. Remove false positives where the tool has over-redacted. This review step is where clinical knowledge matters. A data manager who knows the trial protocol will catch edge cases that pattern detection misses.
Stage 5: Verify the output. Once redactions are applied, scroll through the completed document. Attempt to select text beneath any redaction marks to confirm the underlying data has been removed rather than merely covered. A document where text remains selectable under a black box has not been permanently redacted.
Stage 6: Log and deliver. Record every redaction decision in an audit log: identifier type, document reference, timestamp, and the name of the reviewer who approved the output. Deliver the redacted pack through a secure channel. Retain the original, unredacted documents in your secure trial master file.
Common Mistakes That Create Compliance Risk
-
Incomplete redaction of initials. Full names are redacted but initials remain. If the subject code links a set of initials to a specific patient in the trial register, those initials are identifying. Both must be redacted.
-
Using overlay tools instead of permanent redaction. Applying a black box in a standard PDF editor places a visual layer over the text. The underlying content remains in the document structure. Anyone can remove the annotation and read the original. This is not redaction.
-
No audit trail. If an inspector asks how patient identifiability was protected before documents were shared with the sponsor, "we used Adobe to apply black boxes" is not an auditable response. You need a documented log of what was redacted, by whom, and when.
-
Working on originals. Any process that modifies source documents rather than working on copies creates chain-of-custody problems and may compromise the integrity of the trial record.
-
Inconsistent standards across staff. When multiple coordinators redact documents manually across a large trial, redaction quality varies. Different team members may apply different thresholds to the same identifier types.
How Purpose-Built Software Handles This at Scale
Manual redaction is workable for a small monitoring visit with a handful of subjects. It does not scale.
A site running 20 concurrent trials may receive sponsor audit requests covering hundreds of subjects and thousands of pages. A CRO preparing for a regulatory inspection may need to process five years of records across multiple investigator sites. In these environments, manual redaction creates both a throughput problem and a consistency problem.
Purpose-built redaction software addresses both.
Automated PII detection scans for structured and unstructured identifiers simultaneously, including NHS numbers and other country-specific identifiers, using both pattern matching and natural language processing for contextual text. Handwritten content is processed through ICR.
Configurable confidence thresholds allow clinical data managers to set the certainty level required before a redaction is automatically applied. Items below the threshold are flagged for human review rather than auto-redacted, maintaining oversight without slowing throughput.
A complete audit log is generated automatically: identifier category, document reference, timestamp, and reviewer. This log is available for sponsor review or regulatory inspection.
Redacted output is generated as a clean copy. The original document is preserved separately and untouched.
Small teams processing high-volume records requests face a compounded version of this challenge. The same automation principles that apply at CRO scale also benefit leaner compliance functions, as covered in How Small Healthcare Teams Can Automate Large-Scale Document Redaction.
VIDIZMO Redactor supports clinical research document redaction, including detection of UK NHS numbers and other country-specific identifiers. For organisations handling healthcare data redaction at scale, explore VIDIZMO Redactor and its capabilities for clinical research workflows.
Conclusion
Redacting clinical trial source documents for sponsor audits is a repeatable process, not an ad hoc task. A documented workflow covering receive, identify, detect, review, verify, log, and deliver produces consistent output, supports regulatory inspection, and protects patient privacy at every step.
The quality of that process depends heavily on the tools used and the documentation maintained. Manual redaction creates gaps. Overlay tools create risk. A logged, verified, automated workflow creates an audit trail.
See how VIDIZMO Redactor supports clinical trial document redaction. Request a demo or explore VIDIZMO's redaction software for clinical research workflows.

About the Author

Jump to
How Small Healthcare Teams Can Automate Large-Scale Document Redaction
How to Redact Documents Before Uploading to AI Tools Like ChatGPT


No Comments Yet
Let us know what you think